Pharmacokinetics
Pharmacokinetics, sometimes abbreviated as PK, (from Ancient Greek pharmakon "drug" and kinetikos "to do with motion"; see chemical kinetics) is a branch of pharmacology dedicated to the determination of the fate of substances administered externally to a living organism. In practice this discipline is applied mainly to drug substances, though in principle it concerns itself with all manner of compounds ingested or otherwise delivered externally to an organism, such as nutrients, metabolites, hormones, toxins, etc.
Pharmacokinetics is often studied in conjunction with pharmacodynamics. Pharmacodynamics explores what a drug does to the body, whereas pharmacokinetics explores what the body does to the drug. Pharmacokinetics includes the study of the mechanisms of absorption and distribution of an administered drug, the rate at which a drug action begins and the duration of the effect, the chemical changes of the substance in the body (e.g. by enzymes) and the effects and routes of excretion of the metabolites of the drug.[1]
Contents |
ADME
Pharmacokinetics is divided into several areas which includes the extent and rate of Absorption, Distribution, Metabolism and Excretion. This is commonly referred to as the ADME scheme. However recent understanding about the drug-body interactions brought about the inclusion of new term Liberation. Now Pharmacokinetics can be better described as LADME.
- Liberation is the process of release of drug from the formulation.
- Absorption is the process of a substance entering the blood circulation.
- Distribution is the dispersion or dissemination of substances throughout the fluids and tissues of the body.
- Metabolism is the irreversible transformation of parent compounds into daughter metabolites.
- Excretion is the elimination of the substances from the body. In rare cases, some drugs irreversibly accumulate in a tissue in the body.
Pharmacokinetics describes how the body affects a specific drug after administration. Pharmacokinetic properties of drugs may be affected by elements such as the site of administration and the concentration in which the drug is administered. These may affect the absorption rate.[2]
Parameters
The following are the most commonly measured pharmacokinetic parameters:[3]
| Variable | Description | Example value | Abbreviation(s) | Formula |
|---|---|---|---|---|
| Dose | loading dose (LD), or steady state/maintenance dose (MD) | 1000 mg |  |
 |
| Volume of distribution | The apparent volume in which a drug is distributed immediately after it has been injected intravenously and equilibrated between plasma and the surrounding tissues. | 25 L | 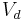 |
 |
| Concentration | initial or steady-state concentration of drug in plasma | 40.0 mg/L |  |
 |
| Biological half-life | The time required for the concentration of the drug to reach half of its original value. | 14 hr |  |
 |
| Elimination rate constant | The rate at which drugs are removed from the body. | 0.05 /hr |  |
 |
| Elimination rate | rate of infusion required to balance elimination | 50 mg/hr |  |
 |
| Area under the curve | The integral of the plasma drug concentration (Cp) after it is administered. | 0.1 mg/mL×hr |  |
 |
| Clearance | The volume of plasma cleared of the drug per unit time. | 1.25 L/hr |  |
 |
| Bioavailability | The fraction of drug that is absorbed. | 1 |  |
![= \frac{[AUC]_{oral}\div dose_{oral}}{[AUC]_{IV}\div dose_{IV}}](/2010-wikipedia_en_wp1-0.8_orig_2010-12/I/68cb4d79503e0c9ed1aec2845aa018e4.png) |
| Cmax | The peak plasma concentration of a drug after oral administration. | 40.0 mg/L |  |
direct measurement |
| Cmin | The lowest (trough) concentration that a drug reaches before the next dose is administered. | 1.0 mg/L |  |
direct measurement |
| edit | ||||
The following graph depicts a typical time course of drug plasma concentration and illustrates the Cmax, Cmin, T1/2 and AUC pharmacokinetic parameters:

Analysis
Pharmacokinetic analysis is performed by noncompartmental (model independent) or compartmental methods. Noncompartmental methods estimate the exposure to a drug by estimating the area under the curve of a concentration-time graph. Compartmental methods estimate the concentration-time graph using kinetic models. Compartment-free methods are often more versatile in that they do not assume any specific compartmental model and produce accurate results also acceptable for bioequivalence studies.
Noncompartmental analysis
Noncompartmental PK analysis is highly dependent on estimation of total drug exposure. Total drug exposure is most often estimated by Area Under the Curve methods, with the trapezoidal rule (numerical differential equations) the most common area estimation method. Due to the dependence on the length of 'x' in the trapezoidal rule, the area estimation is highly dependent on the blood/plasma sampling schedule. That is, the closer your time points are, the closer the trapezoids are to the actual shape of the concentration-time curve.
Compartmental analysis
Compartmental PK analysis uses kinetic models to describe and predict the concentration-time curve. PK compartmental models are often similar to kinetic models used in other scientific disciplines such as chemical kinetics and thermodynamics. The advantage of compartmental over some noncompartmental analyses is the ability to predict the concentration at any time. The disadvantage is the difficulty in developing and validating the proper model. Compartment-free modeling based on curve stripping does not suffer this limitation. The simplest PK compartmental model is the one-compartmental PK model with IV bolus administration and first-order elimination. The most complex PK models (called PBPK models) rely on the use of physiological information to ease development and validation.
Bioanalytical methods
Bioanalytical methods are necessary to construct a concentration-time profile. Chemical techniques are employed to measure the concentration of drugs in biological matrix, most often plasma. Proper bioanalytical methods should be selective and sensitive.
Mass spectrometry
Pharmacokinetics is often studied using mass spectrometry because of the complex nature of the matrix (often blood or urine) and the need for high sensitivity to observe low dose and long time point data. The most common instrumentation used in this application is LC-MS with a triple quadrupole mass spectrometer. Tandem mass spectrometry is usually employed for added specificity. Standard curves and internal standards are used for quantitation of usually a single pharmaceutical in the samples. The samples represent different time points as a pharmaceutical is administered and then metabolized or cleared from the body. Blank or t=0 samples taken before administration are important in determining background and insuring data integrity with such complex sample matrices. Much attention is paid to the linearity of the standard curve; however it is not uncommon to use curve fitting with more complex functions such as quadratics since the response of most mass spectrometers is less than linear across large concentration ranges.[4][5][6]
There is currently considerable interest in the use of very high sensitivity mass spectrometry for microdosing studies, which are seen as a promising alternative to animal experimentation.[7]
Population pharmacokinetics
Population pharmacokinetics is the study of the sources and correlates of variability in drug concentrations among individuals who are the target patient population receiving clinically relevant doses of a drug of interest.[8][9] Certain patient demographic, pathophysiological, and therapeutical features, such as body weight, excretory and metabolic functions, and the presence of other therapies, can regularly alter dose-concentration relationships. For example, steady-state concentrations of drugs eliminated mostly by the kidney are usually greater in patients suffering from renal failure than they are in patients with normal renal function receiving the same drug dosage. Population pharmacokinetics seeks to identify the measurable pathophysiologic factors that cause changes in the dose-concentration relationship and the extent of these changes so that, if such changes are associated with clinically significant shifts in the therapeutic index, dosage can be appropriately modified.
Software packages used in population pharmacokinetics modeling include NONMEM, which was developed at UCSF.
See also
- blood alcohol concentration
- Biological half-life
- bioavailability
- toxicokinetics
- Patlak plot
- Plateau Principle
References
- ↑ pharmacokinetics. (2006). In Mosby's Dictionary of Medicine, Nursing, & Health Professions. Philadelphia, PA: Elsevier Health Sciences. Retrieved December 11, 2008, from http://www.credoreference.com/entry/6686418
- ↑ Kathleen Knights; Bronwen Bryant (2002). Pharmacology for Health Professionals. Amsterdam: Elsevier. ISBN 0-7295-3664-5.
- ↑ AGAH working group PHARMACOKINETICS (2004-02-16). "Collection of terms, symbols, equations, and explanations of common pharmacokinetic and pharmacodynamic parameters and some statistical functions". Arbeitsgemeinschaft für Angewandte Humanpharmakologie (AGAH) (Association for Applied Human Pharmacology). http://www.agah.info/uploads/media/PK-glossary_PK_working_group_2004.pdf.
- ↑ Hsieh Y, Korfmacher WA (June 2006). "Increasing speed and throughput when using HPLC-MS/MS systems for drug metabolism and pharmacokinetic screening". Current Drug Metabolism 7 (5): 479–89. doi:10.2174/138920006777697963. PMID 16787157. http://www.bentham-direct.org/pages/content.php?CDM/2006/00000007/00000005/0004F.SGM.
- ↑ Covey TR, Lee ED, Henion JD (October 1986). "High-speed liquid chromatography/tandem mass spectrometry for the determination of drugs in biological samples". Anal. Chem. 58 (12): 2453–60. doi:10.1021/ac00125a022. PMID 3789400.
- ↑ Covey TR, JB Crowther, EA Dewey, JD Henion (February 1985). "Thermospray liquid chromatography/mass spectrometry determination of drugs and their metabolites in biological fluids". Anal. Chem. 57 (2): 474–81. doi:10.1021/ac50001a036. PMID 3977076.
- ↑ Committee for Medicinal Products for Human Use (CHMP) (2004-06-23). "Position Paper on Non-Clinical Studies to Support Clinical Trials with a Single Microdose" (in en) (PDF). CPMP/SWP/2599/02 Rev 1. European Medicines Agency, Evaluation of Medicines for Human Use. http://www.emea.europa.eu/pdfs/human/swp/259902en.pdf. Retrieved 2008-06-09.
- ↑ Sheiner, L.B.; Rosenberg, B., Marathe, V.V. (1977). "Estimation of Population Characteristics of Pharmacokinetic Parameters from Routine Clinical Data". J. Pharmacokin. Biopharm. 5: 445–79. doi:10.1007/BF01061728.
- ↑ Sheiner, L.B.; Beal, S.L., Rosenberg, B. Marathe, V.V. (1979). "Forecasting Individual Pharmacokinetics". Clin. Pharmacol. Ther. 26 (3): 294–305. PMID 466923.
|
|||||||||||||||||